
Page 3 - B.Bioinformatics and systems biology
P. 3
A Study on the Improvement of the Cryptographic Alogrithm
for Identifying Genetic relatives
1
1
Gi Ju Lee , Minhyeok Jeong , Jong Wha J. Joo 1 *
1 Department of Computer Science and Engineering, Dongguk University-Seoul, 04620 Seoul, Republic of Korea
ABSTRACT
With the advent of high-throughput technology, genetic information is often compared to reveal individual
relationships. Individual genetic information is one of the most important personal information, and efforts are being
made to compare individual genetic information without compromising privacy. Of these, GenoCrypt identifying
genetic relatives without compromising privacy. However, it requires a lot of computing resources. In this study, we
propose a method referred to as AdGenoCrypt while consuming less computing resources.
METHODS
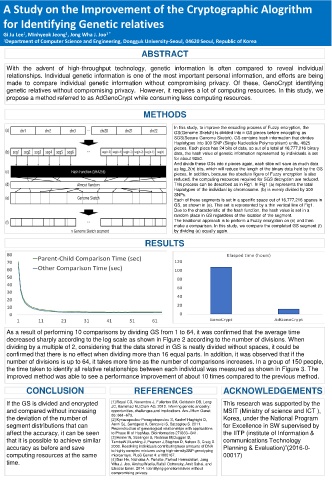
In this study, to improve the encoding process of Fuzzy encryption, the
GS(Genome Sketch) is divided into n GS pieces before encrypting as
SGS(Secure Genome Sketch). GS contains hash information that divides
Haplotypes into 300 SNP (Single Nucleotide Polymorphism) units, 4625
pieces. Each piece has 24 bits of data, so out of a total of 16,777,216 binary
data, the hash value of genetic information represented by individuals is set
for about 9250.
And divide these GSs into n pieces again, each slice will save as much data
as log_2(n) bits, which will reduce the length of the binary data held by the GS
pieces. In addition, because the absolute figure of Fuzzy encryption is also
reduced, the computing resources required for SGS decryption are reduced.
This process can be described as in Fig1. In Fig1 (a) represents the total
Haplotypes of the individual by chromosome. (b) is evenly divided by 300
SNPs.
Each of these segments is set in a specific space out of 16,777,216 spaces in
GS, as shown in (e). This set is represented by a thin vertical line of Fig1.
Due to the characteristic of the hash function, the hash value is set in a
random place in GS regardless of the location of the segment.
The traditional approach is to perform a Fuzzy encryption on (e) and then
make a comparison. In this study, we compare the completed GS segment (f)
by dividing (e) equally again.
RESULTS
As a result of performing 10 comparisons by dividing GS from 1 to 64, it was confirmed that the average time
decreased sharply according to the log scale as shown in Figure 2 according to the number of divisions. When
dividing by a multiple of 2, considering that the data stored in GS is neatly divided without spaces, it could be
confirmed that there is no effect when dividing more than 16 equal parts. In addition, it was observed that if the
number of divisions is up to 64, it takes more time as the number of comparisons increases. In a group of 150 people,
the time taken to identify all relative relationships between each individual was measured as shown in Figure 3. The
improved method was able to see a performance improvement of about 10 times compared to the previous method.
CONCLUSION REFERENCES ACKNOWLEDGEMENTS
If the GS is divided and encrypted [1] Royal CD, Novembre J, Fullerton SM, Goldstein DB, Long This research was supported by the
JC, Bamshad MJ,Clark AG. 2010. Inferring genetic ancestry:
and compared without increasing opportunities, challenges,and implications. Am J Hum Genet MSIT (Ministry of science and ICT ),
the deviation of the number of 86: 661–673. Korea, under the National Program
[2] Kyriazopoulou-Panagiotopoulou S, Kashef Haghighi D,
segment distributions that can Aerni SJ, Sundquist A, Bercovici S, Batzoglou S. 2011. for Excellence in SW supervised by
Reconstruction of genealogical relationships with applications
affect the accuracy, it can be seen to Phase III of HapMap. Bioinformatics 27:i333–i341. the IITP (institute of Information &
that it is possible to achieve similar [3] Homer N, Szelinger S, Redman M,Duggan D, communications Technology
TembeW,Muehling J, Pearson J,Stephan D, Nelson S, Craig D.
accuracy as before and save 2008. Resolving individuals contributingtrace amounts of DNA Planning & Evaluation)”(2016-0-
to highly complex mixtures using high-densitySNP genotyping
computing resources at the same microarrays. PLoS Genet 4: e1000167. 00017)
time. [4] Dan He, Nicholas A. Furlotte, Farhad Hormozdiari, Jong
Wha J. Joo, AkshayWadia, Rafail Ostrovsky, Amit Sahai, and
Eleazar Eskin. 2014. Identifying geneticrelatives without
compromising privacy.